Esclerose Múltipla (EM)


esclerose múltipla
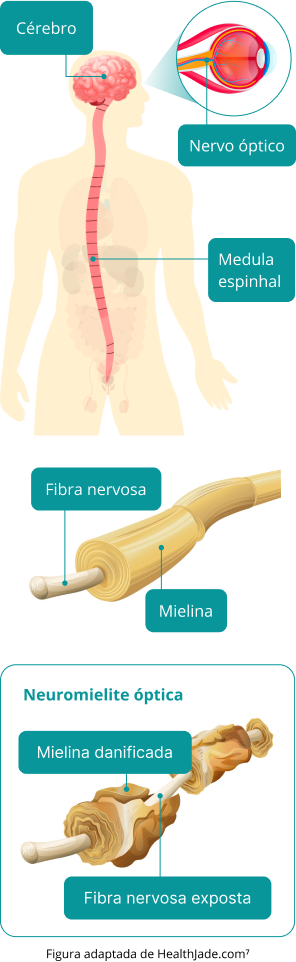
O termo “esclerose múltipla” se refere a várias áreas de cicatrização (esclerose) resultantes da destruição dos tecidos que envolvem os nervos (bainha da mielina) no cérebro e na medula espinhal. Essa destruição denomina-se desmielinização. Às vezes, as fibras nervosas que enviam mensagens (axônio) também são afetadas. Com o tempo, o cérebro pode encolher, pois os axônios são destruídos.
Em todo o mundo, cerca de 2,8 milhões de pessoas têm esclerose múltipla e cerca de 107.000 pessoas são diagnosticadas com esclerose múltipla a cada ano.
Mais comumente, a esclerose múltipla começa entre os 20 e 40 anos de idade, mas pode começar a qualquer momento entre os 15 e 60 anos de idade. De certa forma, ela é mais comum em mulheres. A esclerose múltipla é incomum em crianças.
A maioria das pessoas com esclerose múltipla tem períodos de saúde relativamente boa (remissões), alternando com períodos de piora nos sintomas (surtos ou recaídas). As recaídas podem ser moderadas ou debilitantes. A recuperação durante a remissão é boa, porém muitas vezes incompleta. Assim, a esclerose
Dores locais: nos olhos
Dores circunstanciais: com o movimento dos olhos ou nas costas ao acenar com a cabeça
Nos músculos: dificuldade para caminhar, fraqueza muscular, incapacidade de mudar rapidamente os movimentos, músculos rígidos, problemas de coordenação, rigidez muscular, espasmos musculares ou reflexos hiperativos
No corpo: fadiga, falta de equilíbrio, intolerância ao calor, tontura ou vertigem
No trato urinário: desejo persistente de urinar, incontinência urinária, micção excessiva durante a noite ou retenção urinária
Sensorial: formigamento, formigamento e queimação desconfortável ou redução na sensação de tato
Na visão: perda de visão, visão dupla ou visão embaçada
No sexo: disfunção erétil ou disfunção sexual
No humor: ansiedade ou mudanças de humor
Na fala: dificuldade de fala ou fala arrastada
Também é comum: constipação, dificuldade em engolir, dificuldade em pensar e compreender, dor de cabeça, dormência na língua, dormência no rosto, fraqueza, movimento rápido involuntário dos olhos, privação de sono ou tremor durante movimentos precisos
Apenas para fins informativos. Consulte um médico de confiança para receber orientações adequadas.
Dores circunstanciais: com o movimento dos olhos ou nas costas ao acenar com a cabeça
Nos músculos: dificuldade para caminhar, fraqueza muscular, incapacidade de mudar rapidamente os movimentos, músculos rígidos, problemas de coordenação, rigidez muscular, espasmos musculares ou reflexos hiperativos
No corpo: fadiga, falta de equilíbrio, intolerância ao calor, tontura ou vertigem
No trato urinário: desejo persistente de urinar, incontinência urinária, micção excessiva durante a noite ou retenção urinária
Sensorial: formigamento, formigamento e queimação desconfortável ou redução na sensação de tato
Na visão: perda de visão, visão dupla ou visão embaçada
No sexo: disfunção erétil ou disfunção sexual
No humor: ansiedade ou mudanças de humor
Na fala: dificuldade de fala ou fala arrastada
Também é comum: constipação, dificuldade em engolir, dificuldade em pensar e compreender, dor de cabeça, dormência na língua, dormência no rosto, fraqueza, movimento rápido involuntário dos olhos, privação de sono ou tremor durante movimentos precisos
Apenas para fins informativos. Consulte um médico de confiança para receber orientações adequadas.
Os sintomas da síndrome de Devic, também conhecida como neuromielite óptica, podem variar entre os pacientes e em diferentes fases da doença:
Visão dupla, embaçada, escura ou borrada
Perda parcial do campo de visão
Dor ocular ao movimentar os olhos
Fraqueza ou paralisia nos membros
Alterações na sensibilidade dos membros
Disfunção da bexiga e do intestino
Fadiga
Náuseas e vômitos
Distúrbios do sono
Distúrbios respiratórios
A síndrome de Devic é uma doença autoimune que causa inflamação do nervo óptico e da medula espinhal. O ataque do sistema imunológico é direcionado à aquaporina 4, uma proteína presente nos astrócitos, células de apoio do cérebro, medula espinhal e nervos ópticos.
Não há cura para a doença, mas os tratamentos podem controlar os sintomas e prevenir que eles se repitam. O diagnóstico é feito por meio de exames de sangue, ressonância magnética e outros testes.
Sobre a síndrome anti-MOG
A síndrome anti-MOG é um distúrbio autoimune no qual o sistema imunológico ataca erroneamente a glicoproteína do oligodendrócito da mielina (MOG). Essa proteína está localizada na superfície da mielina, uma camada isolante que protege as células nervosas e ajuda a facilitar a comunicação entre elas. Quando o sistema imunológico ataca a MOG, ele produz anticorpos para a proteína. Os anticorpos normalmente nos protegem de vírus e bactérias, mas os anticorpos para a MOG podem fazer com que a bainha de mielina caia das células nervosas. Esse processo, conhecido como desmielinização, pode impedir que os neurônios transmitam mensagens de forma eficaz e resultar em sintomas neurológicos.
Sintomas da síndrome anti-MOG
Indivíduos com síndrome anti-MOG podem apresentar neurite óptica, mielite transversa e/ou encefalomielite disseminada aguda, dependendo de quais partes do sistema nervoso são afetadas.
Os sintomas da encefalomielite disseminada aguda podem incluir:Coma
Confusão
Sonolência
Febre
Dor de cabeça
Dormência ou formigamento (alterações sensoriais)
Convulsões
Dificuldade em engolir
Caminhada instável (ataxia)
Fraqueza nos braços ou pernas
Os sintomas da neurite óptica podem incluir:Visão turva ou com outros distúrbios
Daltonismo
Dor ocular que piora com o movimento dos olhos
Perda de visão ou visão gravemente prejudicada em um olho
Os sintomas da mielite transversa,
que tendem a se desenvolver rapidamente ao longo de várias horas a várias semanas, podem incluir:Dor nas costas
Disfunção intestinal e da bexiga
Sentindo que algo está enrolado em seu estômago e na parte inferior das costas
Fraqueza ou paralisia dos membros, começando nas pernas e subindo pelo corpo
Distúrbios sensoriais, como dormência ou sensibilidade ao toque
Sintomas da síndrome anti-MOG
Indivíduos com síndrome anti-MOG podem apresentar neurite óptica, mielite transversa e/ou encefalomielite disseminada aguda, dependendo de quais partes do sistema nervoso são afetadas.
Os sintomas da encefalomielite disseminada aguda podem incluir:Coma
Confusão
Sonolência
Febre
Dor de cabeça
Dormência ou formigamento (alterações sensoriais)
Convulsões
Dificuldade em engolir
Caminhada instável (ataxia)
Fraqueza nos braços ou pernas
O tratamento para sintomas agudos de desmielinização envolve a redução da inflamação. Corticosteroides intravenosos são a primeira linha de tratamento. Em casos raros, se esteroides em altas doses não forem eficazes, os pacientes podem passar por tratamento com imunoglobulina intravenosa (IVIG).
Muitos indivíduos com síndrome anti-MOG se recuperarão completamente após o primeiro tratamento e nunca terão recaída. No entanto, alguns pacientes podem apresentar ataques recorrentes e precisar de um plano de tratamento de longo prazo. Isso pode incluir IVIG e medicamentos, como azatioprina, miofenolato mofetil ou rituximabe.
Informações adicionais
Quão comum é a síndrome anti-MOG?
A síndrome anti-MOG é rara. Um estudo de 2019 relatou a incidência da síndrome anti-MOG como 1,6 por 1 milhão de pessoas.
Quem tem síndrome anti-MOG?
A síndrome anti-MOG afeta homens e mulheres igualmente, e o início da doença geralmente ocorre na faixa dos 20 ou 30 anos. Pesquisas sugerem que pessoas brancas podem ser afetadas com mais frequência do que pessoas de outras raças.
Como a síndrome anti-MOG é diagnosticada?
Seu médico procurará sintomas de doenças neuroinflamatórias que podem ocorrer com a síndrome anti-MOG, como neurite óptica, mielite transversa e encefalomielite disseminada aguda.
Os seguintes testes podem ajudar a confirmar a síndrome anti-MOG:Exames de sangue para verificar anticorpos MOG
Avaliação ocular para avaliar inflamação do nervo óptico
Punção lombar para avaliar o líquido cefalorraquidiano (LCR)
Imagem por ressonância magnética (RM) para ver onde ocorreu a inflamação
Um estudo de 2019 relatou a incidência da síndrome anti-MOG como 1,6 por 1 milhão de pessoas
© Barrow Neurological Institute 2025.
Como se faz o diagnóstico? Além da entrevista e do exame físico feitos pelo neurologista, a investigação inicial costuma incluir exames de sangue, ressonância magnética e análise do líquor (material colhido através da punção lombar). O diagnóstico definitivo depende da detecção do anticorpo anti-MOG no sangue.
O que é MOG? M yelin – bainha protetora ao redor dos nervos
O ligodendrócito – uma célula abundante no sistema nervoso central que constrói mielina
G licoproteína – um tipo de molécula de proteína que tem um carboidrato ligado a ela
O primeiro artigo que propôs um protocolo que detecta de forma confiável o anticorpo MOG foi publicado em 2015. Muitos laboratórios ao redor do mundo adotaram o protocolo. Dos pacientes com NMOSD que testam negativo para o anticorpo aquaporina-4, aproximadamente 40% têm anticorpos MOG. A função biológica do MOG ainda não está completamente clara, mas sabemos que ele é um alvo de uma resposta imune aberrante em pessoas com esse transtorno.
Como um de um espectro de distúrbios inflamatórios óptico-espinhais, a MOG compartilha muitas características com a NMOSD. Pacientes com MOG podem apresentar neurite óptica - inflamação dos nervos ópticos, na qual as bainhas de mielina protetoras são danificadas, e/ou mielite transversa - inflamação de uma seção da medula espinhal, na qual a mielina é danificada.
Pacientes com MOG tendem a ser mais jovens do que aqueles com NMOSD, e a doença pode ser monofásica – ocorrendo apenas uma vez – especialmente em crianças. Portanto, nem todos os pacientes com MOG precisam tomar medicamentos imunossupressores por toda a vida. Aqueles com doença multifásica – ocorrendo mais de uma vez – de fato permanecem tomando medicamentos para evitar recaídas. A MOG atinge homens e mulheres igualmente. Mais pessoas com MOG sofrem de neurite óptica do que de mielite transversa. A neurite óptica simultânea de ambos os nervos ópticos é uma característica da doença de anticorpos MOG.
Afinal, o que é neuromielite óptica?
A doença do espectro da neuromielite óptica (NMOSD) é uma condição rara, por isso talvez você ainda não a conheça.
A NMOSD é uma doença rara autoimune, em que o sistema imunológico é ativado de maneira inadequada, e ataca células e tecidos saudáveis do sistema nervoso central (SNC).¹ Nesse processo, o nervo óptico e a medula espinhal sofrem inflamação por conta de uma astrocitopatia (morte de células do SNC, chamadas astrócitos) e secundariamente perdem a bainha de mielina, camada que recobre e protege os neurônios, em um processo chamado de desmielinização inflamatória.²
Descrita pela primeira vez em 1894, era compreendida como um subtipo de esclerose múltipla até 2004, quando graças ao avanço da ciência médica foram observados autoanticorpos específicos e o envolvimento do sistema complemento.²
Em geral, a NMOSD atinge principalmente mulheres entre 30 e 40 anos, mas pode ocorrer em outras idades e, em menor número, em homens. Há uma prevalência maior entre pessoas afrodescendentes e asiáticas.3-5
A NMOSD pode levar à perda da visão e dos movimentos e, se não tratada, pode ser fatal.² As manifestações ocorrem em surtos recorrentes que podem ser debilitantes e difíceis de recuperar.6 A cada surto, pode haver danos neurológicos irreversíveis, e as chances de deficiência ou morte aumentam.
Os sintomas da síndrome de Devic, também conhecida como neuromielite óptica, podem variar entre os pacientes e em diferentes fases da doença:
Visão dupla, embaçada, escura ou borrada
Perda parcial do campo de visão
Dor ocular ao movimentar os olhos
Fraqueza ou paralisia nos membros
Alterações na sensibilidade dos membros
Disfunção da bexiga e do intestino
Fadiga
Náuseas e vômitos
Distúrbios do sono
Distúrbios respiratórios
A síndrome de Devic é uma doença autoimune que causa inflamação do nervo óptico e da medula espinhal. O ataque do sistema imunológico é direcionado à aquaporina 4, uma proteína presente nos astrócitos, células de apoio do cérebro, medula espinhal e nervos ópticos.
Não há cura para a doença, mas os tratamentos podem controlar os sintomas e prevenir que eles se repitam. O diagnóstico é feito por meio de exames de sangue, ressonância magnética e outros testes.
Disfunção intestinal e da bexiga
Sentindo que algo está enrolado em seu estômago e na parte inferior das costas
Fraqueza ou paralisia dos membros, começando nas pernas e subindo pelo corpo
Distúrbios sensoriais, como dormência ou sensibilidade ao toque
Sintomas da síndrome anti-MOG
Indivíduos com síndrome anti-MOG podem apresentar neurite óptica, mielite transversa e/ou encefalomielite disseminada aguda, dependendo de quais partes do sistema nervoso são afetadas.
Os sintomas da encefalomielite disseminada aguda podem incluir:Coma
Confusão
Sonolência
Febre
Dor de cabeça
Dormência ou formigamento (alterações sensoriais)
Convulsões
Dificuldade em engolir
Caminhada instável (ataxia)
Fraqueza nos braços ou pernas
O tratamento para sintomas agudos de desmielinização envolve a redução da inflamação. Corticosteroides intravenosos são a primeira linha de tratamento. Em casos raros, se esteroides em altas doses não forem eficazes, os pacientes podem passar por tratamento com imunoglobulina intravenosa (IVIG).
Muitos indivíduos com síndrome anti-MOG se recuperarão completamente após o primeiro tratamento e nunca terão recaída. No entanto, alguns pacientes podem apresentar ataques recorrentes e precisar de um plano de tratamento de longo prazo. Isso pode incluir IVIG e medicamentos, como azatioprina, miofenolato mofetil ou rituximabe.
Informações adicionais
Quão comum é a síndrome anti-MOG?
A síndrome anti-MOG é rara. Um estudo de 2019 relatou a incidência da síndrome anti-MOG como 1,6 por 1 milhão de pessoas.
Quem tem síndrome anti-MOG?
A síndrome anti-MOG afeta homens e mulheres igualmente, e o início da doença geralmente ocorre na faixa dos 20 ou 30 anos. Pesquisas sugerem que pessoas brancas podem ser afetadas com mais frequência do que pessoas de outras raças.
Como a síndrome anti-MOG é diagnosticada?
Seu médico procurará sintomas de doenças neuroinflamatórias que podem ocorrer com a síndrome anti-MOG, como neurite óptica, mielite transversa e encefalomielite disseminada aguda.
Os seguintes testes podem ajudar a confirmar a síndrome anti-MOG:Exames de sangue para verificar anticorpos MOG
Avaliação ocular para avaliar inflamação do nervo óptico
Punção lombar para avaliar o líquido cefalorraquidiano (LCR)
Imagem por ressonância magnética (RM) para ver onde ocorreu a inflamação
Um estudo de 2019 relatou a incidência da síndrome anti-MOG como 1,6 por 1 milhão de pessoas
© Barrow Neurological Institute 2025.
Como se faz o diagnóstico? Além da entrevista e do exame físico feitos pelo neurologista, a investigação inicial costuma incluir exames de sangue, ressonância magnética e análise do líquor (material colhido através da punção lombar). O diagnóstico definitivo depende da detecção do anticorpo anti-MOG no sangue.
O que é MOG? M yelin – bainha protetora ao redor dos nervos
O ligodendrócito – uma célula abundante no sistema nervoso central que constrói mielina
G licoproteína – um tipo de molécula de proteína que tem um carboidrato ligado a ela
O primeiro artigo que propôs um protocolo que detecta de forma confiável o anticorpo MOG foi publicado em 2015. Muitos laboratórios ao redor do mundo adotaram o protocolo. Dos pacientes com NMOSD que testam negativo para o anticorpo aquaporina-4, aproximadamente 40% têm anticorpos MOG. A função biológica do MOG ainda não está completamente clara, mas sabemos que ele é um alvo de uma resposta imune aberrante em pessoas com esse transtorno.
Como um de um espectro de distúrbios inflamatórios óptico-espinhais, a MOG compartilha muitas características com a NMOSD. Pacientes com MOG podem apresentar neurite óptica - inflamação dos nervos ópticos, na qual as bainhas de mielina protetoras são danificadas, e/ou mielite transversa - inflamação de uma seção da medula espinhal, na qual a mielina é danificada.
Pacientes com MOG tendem a ser mais jovens do que aqueles com NMOSD, e a doença pode ser monofásica – ocorrendo apenas uma vez – especialmente em crianças. Portanto, nem todos os pacientes com MOG precisam tomar medicamentos imunossupressores por toda a vida. Aqueles com doença multifásica – ocorrendo mais de uma vez – de fato permanecem tomando medicamentos para evitar recaídas. A MOG atinge homens e mulheres igualmente. Mais pessoas com MOG sofrem de neurite óptica do que de mielite transversa. A neurite óptica simultânea de ambos os nervos ópticos é uma característica da doença de anticorpos MOG.
Afinal, o que é neuromielite óptica?
A doença do espectro da neuromielite óptica (NMOSD) é uma condição rara, por isso talvez você ainda não a conheça.
A NMOSD é uma doença rara autoimune, em que o sistema imunológico é ativado de maneira inadequada, e ataca células e tecidos saudáveis do sistema nervoso central (SNC).¹ Nesse processo, o nervo óptico e a medula espinhal sofrem inflamação por conta de uma astrocitopatia (morte de células do SNC, chamadas astrócitos) e secundariamente perdem a bainha de mielina, camada que recobre e protege os neurônios, em um processo chamado de desmielinização inflamatória.²
Descrita pela primeira vez em 1894, era compreendida como um subtipo de esclerose múltipla até 2004, quando graças ao avanço da ciência médica foram observados autoanticorpos específicos e o envolvimento do sistema complemento.²
Em geral, a NMOSD atinge principalmente mulheres entre 30 e 40 anos, mas pode ocorrer em outras idades e, em menor número, em homens. Há uma prevalência maior entre pessoas afrodescendentes e asiáticas.3-5
A NMOSD pode levar à perda da visão e dos movimentos e, se não tratada, pode ser fatal.² As manifestações ocorrem em surtos recorrentes que podem ser debilitantes e difíceis de recuperar.6 A cada surto, pode haver danos neurológicos irreversíveis, e as chances de deficiência ou morte aumentam.
Os sintomas da síndrome de Devic, também conhecida como neuromielite óptica, podem variar entre os pacientes e em diferentes fases da doença:
Visão dupla, embaçada, escura ou borrada
Perda parcial do campo de visão
Dor ocular ao movimentar os olhos
Fraqueza ou paralisia nos membros
Alterações na sensibilidade dos membros
Disfunção da bexiga e do intestino
Fadiga
Náuseas e vômitos
Distúrbios do sono
Distúrbios respiratórios
A síndrome de Devic é uma doença autoimune que causa inflamação do nervo óptico e da medula espinhal. O ataque do sistema imunológico é direcionado à aquaporina 4, uma proteína presente nos astrócitos, células de apoio do cérebro, medula espinhal e nervos ópticos.
Não há cura para a doença, mas os tratamentos podem controlar os sintomas e prevenir que eles se repitam. O diagnóstico é feito por meio de exames de sangue, ressonância magnética e outros testes.
 sintomas de problemas no fígado - eranicle/istock
sintomas de problemas no fígado - eranicle/istock

 dinheiro – Créditos: depositphotos.com / joasouza
dinheiro – Créditos: depositphotos.com / joasouza