A esclerose múltipla (EM) é uma doença de provável origem autoimune, na qual o nosso organismo produz inapropriadamente anticorpos contra estruturas dos nossos nervos, levando à inflamação e destruição dos mesmos (leia: O QUE É UMA DOENÇA AUTOIMUNE).
Neste artigo vamos abordar os seguintes pontos sobre a esclerose múltipla:
PUBLICIDADE / PUBLICIDAD
- O que é a esclerose múltipla.
- Quais são as suas causas.
- Quais são as diferenças entre a esclerose múltipla e a síndrome de Guillain-Barré.
- Quais são os sintomas.
- Como é feito o diagnóstico.
- Quais são as opções de tratamento.
- Tratamentos promissores que podem levar à cura da esclerose múltipla.
O que é esclerose múltipla
A esclerose múltipla é uma doença neurológica que ocorre pela destruição da bainha de mielina, substância que recobre os nervos. Portanto, para compreender a esclerose múltipla é preciso saber antes o que é a bainha de mielina.
Todo o nosso sistema nervoso se comunica através de impulsos elétricos. Por exemplo, quando mexemos a nossa mão, só conseguimos fazê-lo porque o nosso sistema nervoso é capaz de enviar um impulso elétrico, que sai do cérebro, caminha pela medula, passa para os nervos periféricos e chega até os músculos da mão, dando ordem para eles se mexerem. Os impulsos também podem seguir o caminho inverso. Todas as sensações que temos do ambientes (temperatura, tato, pressão, dor, etc.) só são percebidas porque as terminações nervosas da pele conseguem captar esses estímulos, enviando-os aos nervos periféricos, medula e, finalmente, cérebro, onde eles serão interpretados.

Esses estímulos elétricos que chegam e saem do cérebro precisam ser transportados entre um neurônio e outro. O fio condutor dos neurônios responsável por esta conexão é chamado de axônio, um prolongamento do próprio neurônio capaz de ligar uma célula nervosa à outra. Como qualquer fio elétrico, os axônios precisam de um isolamento, como se fosse um fio encapado. A substância que fornece esse isolamento e permite a transmissão dos impulsos elétricos é a bainha de mielina.
Na esclerose múltipla, as células nervosas do cérebro e da medula apresentam progressiva destruição de suas bainhas de mielina, fazendo com que os axônios percam a capacidade de transportar os impulsos elétricos. Os neurônios centrais deixam de enviar e de receber estímulos elétricos.
Causas da esclerose múltipla
A esclerose múltipla é uma doença autoimune causada pela destruição da bainha de mielina pelos nossos próprios anticorpos. Não sabemos bem o porquê, mas de uma hora para outra o nosso organismo passa a tratar a bainha de mielina presente nos axônios do sistema nervoso central como uma estrutura estranha, como se fosse um vírus ou bactéria. O sistema imune passa, então, a atacar a bainha de mielina destes neurônios, destruindo-a progressivamente. Imagina-se que a origem da esclerose múltipla possa estar relacionada a desarranjos do sistema imunológicos que surgem após algumas doenças virais, como, por exemplo, a mononucleose (leia: MONONUCLEOSE – DOENÇA DO BEIJO).
Diferenças entre esclerose múltipla e síndrome de Guillain-Barré
A esclerose múltipla e a síndrome de Guillain-Barré são doenças semelhantes na medida em que ambas tem origem autoimune e ocorrem por ataques à bainha de mielina dos nervos (leia: O QUE É A SÍNDROME DE GUILLAIN-BARRÉ?). A diferença é que no Guillain-Barré os nervos acometidos são os do sistema nervoso periférico (nervos fora da medula), enquanto que na esclerose múltipla são os nervos do sistema nervoso central (medula e cérebro) que sofrem desmielinização. Essa pequena diferença é importantíssima no prognóstico final, uma vez que os nervos periféricos têm capacidade de se regenerar, enquanto que os neurônios e axônios do cérebro e da medula não.
Fatores de risco para esclerose múltipla
A esclerose múltipla normalmente se manifesta pela primeira vez entre os 20 e 40 anos de idade. É duas vezes mais comum em mulheres do que em homens, e três vezes mais comum em pessoas que tenham algum familiar acometido pela doença. A esclerose múltipla ocorre com mais frequência em caucasianos (brancos) do que em afrodescendentes ou asiáticos.
Aparentemente, um dos fatores de risco para o surgimento da esclerose múltipla é a infecção pelo vírus Epstein Barr, causador da mononucleose. Imagina-se que o vírus possa ter proteínas semelhantes às da bainha de mielina, fazendo com que os anticorpos tenham dificuldade de distingui-las. É importante frisar que a imensa maioria dos pacientes que tiveram contato com o vírus Epstein Barr não desenvolvem a esclerose múltipla, o que sugere que mais de um fator seja necessário para o surgimento da doença.
Pacientes portadores de outras doenças autoimunes, como tireoidite de Hashimoto (leia: HIPOTIREOIDISMO | TIREOIDITE DE HASHIMOTO), diabetes mellitus tipo 1 (leia: O QUE É DIABETES?) ou doença de Crohn (leia: DOENÇA DE CROHN | RETOCOLITE ULCERATIVA) também apresentam maior risco de desenvolverem esclerose múltipla.
Nos últimos anos tem-se dado muita atenção à relação entre níveis de vitamina D e a esclerose múltipla. Sabemos que a doença é menos comum em áreas tropicais, onde a incidência solar anual é maior e a produção de vitamina D pela pele é mais intensa. Estudos sugerem que níveis adequados de vitamina D podem ser um fator de proteção contra a esclerose múltipla (leia: VITAMINA D | Deficiência e suplementos).
Sintomas da esclerose múltipla
Os sinais e sintomas da esclerose múltipla dependem de quais pontos do sistema nervoso são afetados. Não existe um sintoma típico que feche o diagnóstico de esclerose múltipla, porém, alguns deles são muito sugestivos:
PUBLICIDADE / PUBLICIDAD
– Neurite óptica: normalmente se apresenta como um dor aguda em um dos olhos, que piora com o movimento ocular. Esta dor costuma vir associada a graus variáveis de perda visual, geralmente no centro do campo visual. O paciente pode também apresentar visão dupla ou borrada. Nistagmo (discreto movimento involuntário dos olhos) é um achado comum.
O acometimento dos dois olhos ao mesmo tempo é incomum na esclerose múltipla e costuma indicar outra doença neurológica.
– Sintomas sensoriais: formigamento e dormências, principalmente nos membros, ocorrendo em um lado do corpo de cada vez, são sintomas muito comuns da esclerose múltipla e aparecem em quase 100% dos casos ao longo do curso da doença.
– Fenômeno de Lhermitte: sensação de choque elétrico que se irradia pela coluna vertebral, desencadeado por movimentos da cabeça e do pescoço é chamado de Fenômeno de Lhermitte. É um sintoma típico da esclerose múltipla, mas pode também ocorrer em outras doenças neurológicas.
– Tonturas e vertigens: até 50% dos pacientes com EM podem apresentar tonturas (leia: TONTURA E VERTIGEM | Causas e sintomas). Este sintoma geralmente surge em pacientes com acometimento da face pela doença, como dormências e alterações oculares e auditivas.
– Sintomas motores: tremores, alterações na marcha, diminuição de força muscular e paralisias dos membros ocorrem por lesão dos neurônios da medula. A perda de força é inicialmente unilateral, mas torna-se bilateral em fases avançadas. O acometimento dos membros inferiores é tipicamente mais intenso do que nos membros superiores.
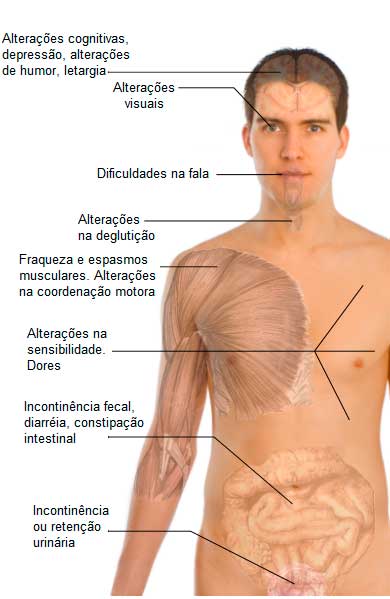
– Incapacidade de controlar a bexiga e os intestinos: A lesão dos nervos da medula além de causar fraqueza muscular nos membros inferiores, também pode provocar uma perda do controle dos esfincteres anal e da bexiga, provocando incontinência fecal e urinária
A esclerose múltipla se manifesta alternando períodos de ataques com remissões. O doente apresenta sintomas agudos que duram dias a semanas, e depois somem, podendo deixar ou não sequelas. O paciente permanece assintomático até um segundo ataque, que também desaparece. Conforme os ataques vão se acumulando, eles ficam cada vez mais agressivos e as sequelas vão se somando, de modo que o paciente vai ficando progressivamente pior ao final de cada exacerbação.
A sobrevida dos pacientes com esclerose múltipla atualmente é de 30 a 40 anos. Pacientes que após 10 ou 15 anos de doença possuem pouca ou nenhuma sequela são aqueles com melhor prognóstico, apresentando maior tempo e qualidade de vida.
Evolução da esclerose múltipla
A esclerose múltipla pode ter apresentações distintas entre os paciente. Há alguns padrões de comportamento são bem conhecidos.
PUBLICIDADE / PUBLICIDAD
1. Esclerose Múltipla Remitente Recorrente (EMRR) ou surto remissão.
Esta forma de esclerose múltipla é caracterizada por surtos de início súbito, mas de curta duração, seguidos por recuperação completa (ou parcial com sequelas mínimas). Não há progressão da doença fora dos períodos de surtos, e o paciente pode ficar meses ou anos sem sinais da da esclerose múltipla. Este padrão de EM é responsável por 85 a 90% dos casos iniciais. No entanto, a maioria dos pacientes com EMRR irá eventualmente entrar numa fase progressiva da doença, chamada Esclerose Múltipla Secundaria Progressiva (EMSP).
2. Esclerose Múltipla Secundaria Progressiva (EMSP)
A esclerose múltipla secundária progressiva ocorre quando há agravamento da forma Esclerose Múltipla Remitente Recorrente (EMRR), geralmente 15 a 20 anos após o início da doença. Nesta forma as crises se tornam mais frequentes e as sequelas começam a se acumular. O paciente agora pode evoluir com piora dos sintomas mesmo sem haver crises agudas.
3. Esclerose Múltipla Primária Progressiva (EMPP)
A esclerose múltipla progressiva primária é caracterizada pela progressão rápida da doença desde fases iniciais. O paciente pode não ter surtos, mas vai acumulando sintomas e sequelas progressivamente. Este tipo tem prognóstico pior e representa cerca de 10 por cento dos casos. Surge habitualmente em pacientes que desenvolvem EM após os 40 anos.
Diagnóstico da esclerose múltipla
Não existe um exame único que estabeleça o diagnóstico da esclerose múltipla. O diagnóstico é feito através da interpretação dos sintomas e de alguns exames completares. Os exames mais usados para a elucidação do quadro são a ressonância magnética nuclear do sistema nervoso central, a análise do líquido cefalorraquidiano, obtido através da punção lombar, e o teste de potencial evocado, que consiste na avaliação da resposta do organismo a pequenos choques elétricos, que estimulam nervos periféricos da visão ou dos músculos.
Tratamento da esclerose múltipla
Infelizmente, ainda não existe um tratamento amplamente disponível que seja capaz de curar a esclerose múltipla. Há tratamentos experimentais promissores, conforme veremos no tópico a seguir, mas estes ainda estão em fases de testes e não fazem parte, ainda, do tratamento habitual da esclerose múltipla.
Como trata-se de uma doença de origem imunológica, o tratamento da EM baseia-se em drogas que ajam no sistema imunológico. A terapia é divida em tratamento nas crises e tratamento durante a remissão.
Tratamento das crises de esclerose múltipla
Os corticoides são as drogas mais usadas durante os surtos (leia: PREDNISONA E CORTICOIDES | efeitos colaterais). O tratamento é chamado de pulsoterapia e consiste na administração de doses elevadas de corticoides (habitualmente metilprednisolona) por via venosa durante 5 dias.
PUBLICIDADE / PUBLICIDAD
Nos casos de surto grave, com pouca resposta aos corticoides, indica-se a realização da plasmaferese, um procedimento parecido com a hemodiálise, que serve para limpar o sangue dos anticorpos danosos (leia: ENTENDA COMO FUNCIONA A PLASMAFÉRESE).
Tratamento da esclerose múltipla durante a remissão
Já existem drogas para tratar os pacientes fora das crises, visando reduzir as sequelas e a ocorrência de novos surtos. Este tratamento, chamado modificador de doença, é especialmente eficaz nos casos de Esclerose Múltipla Remitente Recorrente (EMRR). Ele não cura a EM, mas melhora muito o seu prognóstico.
As drogas mais usadas atualmente são o interferon beta (Avonex, Rebif ou Betaseron) e o acetato de glatiramer (Copaxone). No Brasil estes medicamentos são distribuídos gratuitamente pelo governo.
Nos casos graves, com pouca resposta ao interferon e ao glatiramer, ainda existe a opção pelo tratamento com natalizumabe (tysabri) ou pulsoterapia mensal com corticoides.
Pacientes com deficiência de vitamina D devem receber suplementos para normalizar seus níveis. Estudos sugerem que a vitamina D ajuda no controle das crises da esclerose múltipla.
Tratamentos promissores da esclerose múltipla
O transplante autólogo de medula óssea tem se mostrado em estudos preliminares uma opção muito promissora para que finalmente tenhamos uma cura da esclerose múltipla.
O tratamento é feito da seguinte forma: os médicos coletam e congelam as células-tronco da medula óssea, que estão em um estágio tão inicial de desenvolvimento, que ainda não adquiriram os defeitos que provocam a EM. A seguir, o sistema imunológico defeituoso do paciente é destruído com quimioterapia. O paciente fica completamente depletado das suas células de defesa. As células-tronco previamente coletadas são então re-infundidas no sangue de forma a repopular o sistema imunológico.
Apesar da agressividade do tratamento e dos riscos de morte durante a fase em que o paciente encontra-se com o sistema imunológico destruído, os resultados têm sido animadores. A grande maioria dos pacientes submetidos a esse tratamento estão livres da doença e muitos que tinham sequelas graves voltaram a ser indivíduos plenamente ativos. É importante destacar que os estudos foram feitos com poucos pacientes e houve casos de mortes por infecção após a fase da quimioterapia.
Que o tratamento funciona, parece não haver dúvidas, a questão agora é torná-lo mais seguro para que ele possa ser indicado para um número grande de pacientes.
