O que é a doença de Kawasaki
A doença de Kawasaki (DK), chamada inicialmente de síndrome dos nódulos linfáticos mucocutâneos, foi descrita no Japão, em 1967, pelo Dr. Tomisaku Kawasaki, que relatou 50 casos de crianças com quadro de febre, erupções a pele, conjuntivite, aumento dos linfonodos do pescoço, inflamação dos lábios e da língua e edema das mãos e dos pés.
A doença foi inicialmente considerada uma enfermidade benigna, pois ela curava-se espontaneamente após cerca de 12 dias, mesmo quando nenhum tratamento havia sido administrado. Porém, conforme novos casos foram sendo descritos, verificou-se que até 25% das crianças não tratadas desenvolviam sequelas cardíacas e cerca de 2% evoluíam para o óbito.
PUBLICIDADE / PUBLICIDAD
A doença de Kawasaki é uma vasculite* que pode provocar lesão em veias e artérias de pequeno e médio calibres em qualquer parte do corpo, mas que atinge de forma mais pronunciada as artérias coronarianas (artérias que nutrem o músculo cardíaco).
* Se você quiser mais explicações sobre o que é uma vasculite, acesse o link: VASCULITE – Causas, Tipos, Sintomas e Tratamento.
A DK é uma doença que ocorre predominantemente em crianças até os 5 anos de idade (80 a 90% dos casos). Apesar de existirem casos em todos os países e em todas as etnias, essa forma de vasculite é muito mais comum nas crianças de origem asiática, principalmente nas japonesas.
Enquanto nos países ocidentais a incidência anual da doença de Kawasaki é baixa, com apenas 10 a 20 casos descritos por cada 100.000 crianças de até 5 anos, no Japão, a incidência é alta, chegando a 250 casos por ano para cada 100.000 crianças. Cerca de 1% das crianças japonesas desenvolvem a doença de Kawasaki ao longo dos primeiros 5 anos de vida.
A doença de Kawasaki no adulto é bastante rara, havendo apenas 100 casos publicados em todo mundo entre 2010 e 2017.
Causas da doença de Kawasaki
A origem da doença de Kawasaki ainda é desconhecida. Estudos epidemiológicos e imunológicos sugerem que o gatilho para a cascata de eventos que acaba por provocar o quadro de vasculite possa ter origem infecciosa. No entanto, a gênese da doença não parece ser assim tão simples e fatores auto-imunes e genéticos também parecem ser necessários para a doença surgir.
A hipótese mais aceita atualmente é a de que um agente infeccioso, seja um vírus ou uma bactéria, possa ativar o sistema imunológico de crianças geneticamente predispostas, provocando uma reação auto-imune contra os vasos sanguíneos (para entender o que é uma doença auto imune, leia: DOENÇAS AUTOIMUNES – Causas, Sintomas e Tratamento).
O mais provável é que esse germes que desencadeiam a doença de Kawasaki provoquem uma infecção assintomática ou pouco sintomática antes da vasculite surgir, motivo pelo qual é difícil estabelecer com certeza a relação da DK com uma infecção prévia.
Alguns dados epidemiológicos importantes dão suporte à teoria da origem infecciosa, entre eles:
- A doença de Kawasaki é caracterizada por um exantema febril com inflamação dos gânglios linfáticos e da mucosa da boca, manifestações que são semelhantes às de várias doenças contagiosas infantis, tais como sarampo e escarlatina, por exemplo.
- Existe um aumento sazonal na incidência de DK no inverno e no verão, comportamento que é semelhante ao de várias infecções virais.
- A doença de Kawasaki ocorre frequentemente em surtos, acometendo uma determinada população em uma restrita área geográfica.
- No Japão observou-se que os irmãos das crianças com DK apresentam maior risco de também desenvolver a doença, o que geralmente ocorre dentro de uma semana após o início dos sintomas na primeira criança.
- A doença é comum em crianças menores de cinco anos, mas é rara naqueles menores de seis meses. Esse fato pode ser explicado pela presença de anticorpos maternos ainda circulantes no organismo do bebê nos primeiros meses de vida, o que impediria a sua contaminação pelos germes desencadeadores.
Entre os vírus e bactérias que podem desencadear a doença de Kawasaki, alguns agentes suspeitos são:
- Parvovírus B19.
- Meningococo.
- Mycoplasma pneumoniae.
- Klebsiella pneumoniae.
- Adenovírus.
- Citomegalovírus.
- Vírus Parainfluenza.
- Rotavírus.
- Vírus do sarampo.
- Vírus de Epstein Barr.
- Vírus linfotrópico humano.
- Rickettsias.
Sintomas da doença de Kawasaki
O quadro clínico da doença de Kawasaki é habitualmente dividido em 3 fases: aguda, subaguda e convalescência, conforme o gráfico abaixo ilustra.
PUBLICIDADE / PUBLICIDAD
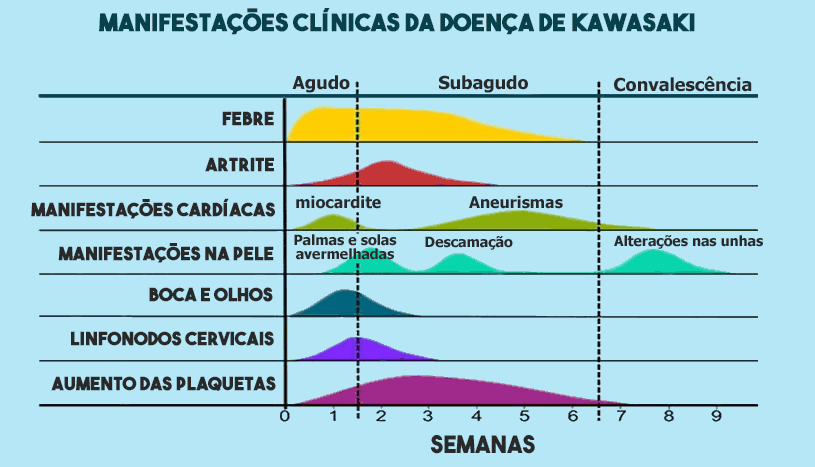
1. Fase aguda
Febre
Febre alta, acima de 38,5ºC, de início súbito e que responde mal aos antipiréticos, é o primeiro e o mais comum sinal da doença de Kawasaki. Se não for tratada, a febre costuma se prolongar por vários dias, às vezes até semanas.
Conjuntivite
Poucos dias após o surgimento da febre, 90% das crianças começam a apresentar conjuntivite bilateral, que costuma provocar vermelhidão mais intensa na parte lateral dos olhos, com pouca ou nenhuma secreção amarelada. 70% dos pacientes com acometimento ocular desenvolvem também uveíte anterior, que é a inflamação da íris ao redor da pupila.
Mucosite
Mais ou menos na mesma época em que surge a conjuntivite, o paciente também costuma desenvolver mucosite, que é a inflamação da mucosa da boca. Os lábios ficam avermelhados e rachados, enquanto a língua fica inflamada e com as papilas bem evidentes, adquirindo um aspecto típico chamado de “língua em morango”, conforme ilustrado na imagem que abre o texto.
Ao contrário da conjuntivite, que ocorre em praticamente todos os casos, a mucosite pode não estar presente em todos os pacientes ou pode ser muito branda, passando quase despercebida.
Rash
Também nos primeiros dias de febre, em cerca de 70% dos pacientes costuma surgir um rash de pele na região genital, perianal e no tronco.
Linfadenite cervical
PUBLICIDADE / PUBLICIDAD
A linfadenite cervical (aumento dos linfonodos no pescoço) costuma estar presente em 25 a 50% dos casos. A maioria dos pacientes apresenta um único linfonodo aumentado na região anterior do pescoço.
Artrite
A artrite costuma aparecer em apenas 10% dos pacientes. As articulações habitualmente mais acometidas são os tornozelos, joelhos e quadril.
Manifestações cardíacas
A miocardite (inflamação do músculo cardíaco) e a pericardite (inflamação do pericárdio) costumam ser a complicações que surgem na fase aguda da doença de Kawasaki. Arritmias e lesões nas válvulas cardíacas também podem surgir.
Ao ecocardiograma, alguns pacientes com miocardite já apresentam nos primeiros dias sinais iniciais de insuficiência cardíaca.
Mãos e pés
Vermelhidão e inchaço nas nas palmas das mãos e na planta dos pés costumam ser a última manifestação a surgir na fase aguda. A lesão nos pés pode fazer com que a criança tenha dificuldade para andar.
Outros sinais e sintomas
Nos primeiros 10 dias de doença, o paciente pode apresentar também:
PUBLICIDADE / PUBLICIDAD
- Diarreia, vômitos ou dor abdominal – 61%.
- Irritabilidade – 50%.
- Vômitos isoladamente – 44%.
- Perda do apetite – 37%.
- Tosse – 35%.
- Dor nas articulações (sem sinais de artrite) – 15%.
2. Fase subaguda
A fase subaguda inicia-se quando a febre desaparece, fato que costuma ocorrer ao redor do 10º dia, e dura até a 4ª ou 6ª semana. O não desaparecimento da febre após 2 semanas costuma ser um sinal de mau prognóstico, pois está mais associado a complicações cardíacas.
A manifestação característica da fase subaguda é a descamação dos dedos, que habitualmente começa nas extremidades e vai se alastrando por todas as mãos e pés. Outro achado típico é o surgimento de aneurismas nas artérias coronarianas, provocados pela inflamação dos vasos sanguíneos.
Sinais e sintomas iniciados na fase aguda, tais como vômitos, diarreia, irritabilidade e dor articular, podem ainda estar presentes nessa fase seguinte da doença.
Laboratorialmente, o paciente apresenta uma grande elevação na contagem de plaquetas (trombocitose), que pode ultrapassar o valor de 1 milhão de células por microlitro, aumentando muito o risco de formação de trombos nos vasos sanguíneos (trombose).
Os valores de VHS e de proteína C reativa (PCR) também costumam estar bem elevados.
3. Fase de convalescência
A fase de convalescência é marcada pela resolução completa dos sintomas da doença, fato que geralmente acontece no prazo de 3 meses.
Durante este estágio, podem surgir sulcos transversais nas unhas, chamadas de linhas de Beau.
Na fase de convalescência, os pequenos aneurismas da artéria coronária tendem a se resolver espontaneamente. Já os aneurismas maiores, por sua vez, podem se expandir e provocar infarto do miocárdio.
Critérios diagnósticos
Não existe um exame laboratorial ou de imagem que sozinho consiga estabelecer o diagnóstico da doença de Kawasaki. Sendo assim, o diagnóstico, em geral, é feito através da avaliação conjunta da história epidemiológica, dos sintomas e dos resultados dos exames complementares.
PUBLICIDADE / PUBLICID
Os critérios diagnósticos estabelecidos pelo Dr. Tomisaku Kawasaki em 1967 ainda são utilizados hoje em dia.
Para que uma criança de até 5 anos tenha o diagnóstico da DK, ela precisa apresentar um quadro de febre por pelo menos 5 dias, associado a pelo menos quatro dos seguintes cinco achados físicos:
- Conjuntivite bilateral.
- Inflamação da mucosa oral.
- Inflamação e edema de mãos e pés.
- Rash característico.
- Linfadenopatia cervical (pelo menos um linfonodo maior que 1,5 cm de diâmetro).
O ecocardiograma é um exame que não faz parte dos critérios diagnósticos, mas é útil para identificar precocemente as complicações cardíacas.
Tratamento da doença de Kawasaki
O tratamento precoce da doença de Kawasaki é essencial para reduzir o risco de complicações.
A administração de imunoglobulina intravenosa em dose única ainda nos primeiros 10 dias doença é a principal medida terapêutica.
Se o diagnóstico não tiver sido feito nos primeiros 10 dias, mas o paciente ainda apresentar sinais de inflamação sistêmica ativa, tais como febre alta e PCR elevada, a imunoglobulina ainda pode ser administrada, mesmo após o 10º dia de doença.
A aspirina em doses elevadas costuma ser utilizada na fase aguda, pois ela além de ter efeito anti-inflamatório e antipirético também inibe a ação das plaquetas, diminuindo o risco de trombose.
A taxa de mortalidade dos pacientes tratados é baixa (0,1 a 0,3%). Os raros casos fatais ocorrem quando há envolvimento cardíaco grave, que geralmente resultam em infarto do miocárdio, arritmias ou ruptura de aneurisma. Esses casos geralmente ocorrem quando o diagnóstico não é feito precocemente e o tratamento não é administrado nos primeiros 10 dias de doença.